本篇说明
本篇论文是 弗罗里达国际大学 的研究人员于 2023 年 8 月 3 日发表在
《Nature Machine Intelligence》 上的一篇研究论文,该团队利用以 Transformer 架构为基础的 ViT(Vision in Transformer) 模型进行 蛋白质-蛋白质结合界面(PPI) 的打分评估,能够对天然蛋白质复合物和不正确构象进行准确区分。具体来说,研究人员参考先前工作(i.e. MaSIF)将蛋白质结合界面转换成二维图像的集合,其中每一张图像对应着界面的几何或生化特性,每一个像素代表相应的特征值,利用 ViT 模型进行界面结合分数评估。同时作者也结合生物物理前验知识,进行了 混合能量项、多注意力模块、对比学习 的多项改进增强,在三个基准测试集上取得了 SOTA 的效果。
相关资料
 bioRxiv
bioRxiv 补充材料
补充材料 测试数据
测试数据 代码
代码蛋白质蛋白质相互作用
背景与现状
- AlphaFold 几乎解决了单体蛋白质结构预测的问题,下一挑战便是蛋白质复合物的结构预测。
- 传统意义上,给定结构可用的单体蛋白质,可以使用分子对接工具(e.g. FireDock、HDock )预测其结合的蛋白质复合物,形成稳定的合理复合构象,从而加速药物、疫苗的设计。然而许多拥有良好结合分数的预测构象,在湿实验中未能成功确认,传统工具还存在较大的不足。
- 对接工具的使用往往依赖于准确的打分函数。第一阶段:预测多个候选构象;第二阶段:对对接构象进行打分排名。通常需要设计数量较多(100个)的候选构象,计算时间长,且常常预测自然界中不相互作用的分子的结合位点,导致假阳率较高。
创新点
- 提出了一种利用视觉模型( ViT )对蛋白质结合界面进行评估打分的模型。
- 引入多注意力轴机制,对不同特征类型分别通过 ViT 进行潜在表示,最后通过 Transformer 在潜在空间中组合,提出了 Multi-Attention 模块。
- 结合 物理先验 知识,提出了 ViT-Hybrid 模块,引入额外的能量项。
- Multi-Attention 与 ViT-Hybrid 的机制对蛋白质结合界面评分提供了有关几何生化特征和界面图的 可解释性 。
- 使用 对比小批量训练 ,每次使用同一复合物的正负样本批次,引入新的损失函数(监督对比损失、边际排序损失、二元交叉熵损失)在嵌入空间上学习 自然对接姿态(正类) 和 错误对接姿态(负类) 之间的差异,也就是使用了 原型学习、对比学习。
PIsToN 训练数据处理
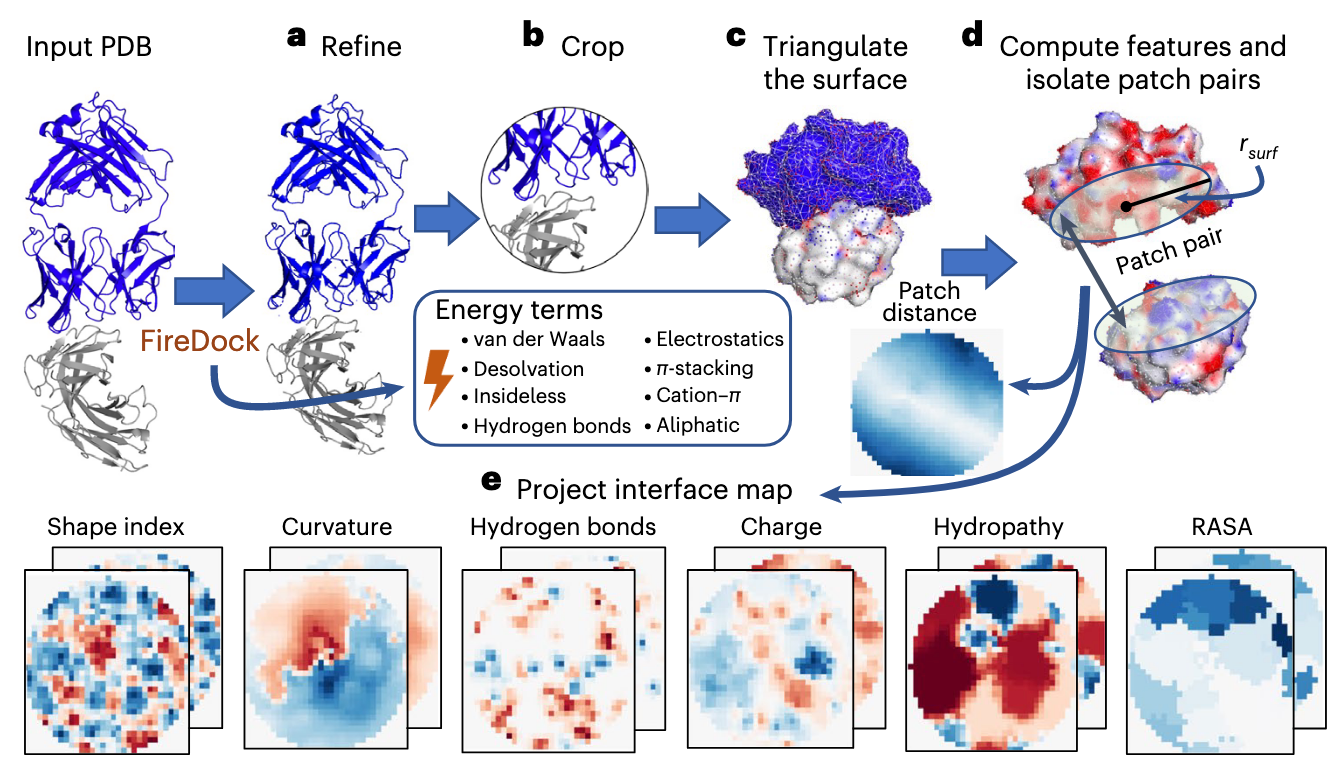
有关将结合界面的几何、生化特征转换为二维的特征图的方法早在 MaSIF 中就已经被使用,本文使用的就是先前相关工作中使用的转换方法。我们的输入是一个复合物结构的 PDB 格式文件 ,依次经过了 Refine、Crop、Triangle、Patch、Project 操作转换为多通道的 特征图(Feature Map) 。
Refine
本步中使用 FireDock 对蛋白质结构进行 Refine ,对其结构进行了 50 个蒙特卡洛循环增强其侧链的灵活性,并将计算后的 Refine 结构的 结合自由能 当作物理先验项保留下来作为输入。其中保留下来的能量项包括:
| 标号 | 特征 | 能量项 |
|---|---|---|
| 00 | 范德华力(van der Waals) | aVdW、rVdW |
| 01 | 脱溶剂化能(desolvation) | ACE |
| 02 | 嵌入性(insideness) | inside |
| 03 | 氢键和二硫键(hydrogen bonds and disulfide bonds) | hb |
| 04 | 静电作用(electrostatics) | aElec、rElec、laElec、lrElec |
| 05 | 相互作用( stacking) | piS |
| 06 | 阳离子 (cation - ) | catpiS |
| 07 | 脂肪相互作用(aliphatic interactions) | aliph |
| 08 | 总能量 | glob |
总共可以分为 8 类,共 13 个能量项。
Crop
对蛋白质复合物进行裁剪。具体来说,定义与对方蛋白原子距离在 之内的为接触点,筛选出所有的接触点之后,计算接触点的几何中心;接着以接触中心为球心,裁剪掉距离该中心超过 (文章中尝试了 、、) 半径以外的区域。
Triangle
这一步将裁剪下来的复杂 3D 蛋白质结构进行三角化,构建由顶点组成的三角网格,并重新缩放到 1 的颗粒度。
Patch
接着,在三角化的结合界面上,计算两个结合蛋白相互作用表面上一对 Patches 的特征。
- Step1:在三角化的蛋白质表面上,计算距离相互作用中心的 测地线距离 在 之内的顶点集合,也就是 Patch 。
- Step2:计算每一个 Patch 表面 上的相关几何、生化特征。
- Step3:再由两个蛋白质表面的点之间的欧几里得距离确定 Patch Distance 的一个几何特征。
- Step4:使用 DSSP v2.365 计算了 Patch 上的 相对可接近表面积。
最终,得到了 两个表面上 的几何生化特征和 一个表面之间 的几何特征。
Project
利用 多维缩放算法 将 Patch 上的表面点投影到二维图上,同时近似保持 Patch 上两点之间的测地线距离,最终形成了大小为 的 2D 网格。其中每一个像素值的强度 是由它周围最近的 4 个蛋白质表面点加权平滑得到的。
特征通道
经过上述操作之后,从输入的 PDB 结构可以得到 13 个通道的特征图。
| 通道 | 特征 |
|---|---|
| 11 | 形状索引 * 2 |
| 12 | 曲率 * 2 |
| 13 | 氢键 * 2 |
| 14 | 带电性 * 2 |
| 15 | 疏水性 * 2 |
| 16 | 相对可接近表面积 * 2 |
| 17 | 界面原子间距离 * 1 |
特征融合
作者提出了 Multi-Attentoin 的机制,通过将不同的特征图和能量项分成 5 组,通过 Hybrid 的方法分别进行潜在表征,最后通过 Transformer 融合嵌入。在代码中可以了解到,作者将特征图和能量项通过如下方式结合:
| 通道 | 特征图 | 能量项 |
|---|---|---|
| 形状互补 | 形状索引 * 2、 曲率 * 2、界面原子间距离 * 1 | aVdW、rVdW、inside |
| 相对可接近表面积 | 相对可接近表面积 * 2、界面原子间距离 * 1 | ACE |
| 氢键作用 | 氢键 * 2、界面原子间距离 * 1 | hb |
| 电性 | 带电性 * 2、界面原子间距离 * 1 | aElec、rElec、laElec、lrElec、piS、catpiS、aliph |
| 疏水性 | 疏水性 * 2、界面原子间距离 * 1 |
PIsToN 模型架构
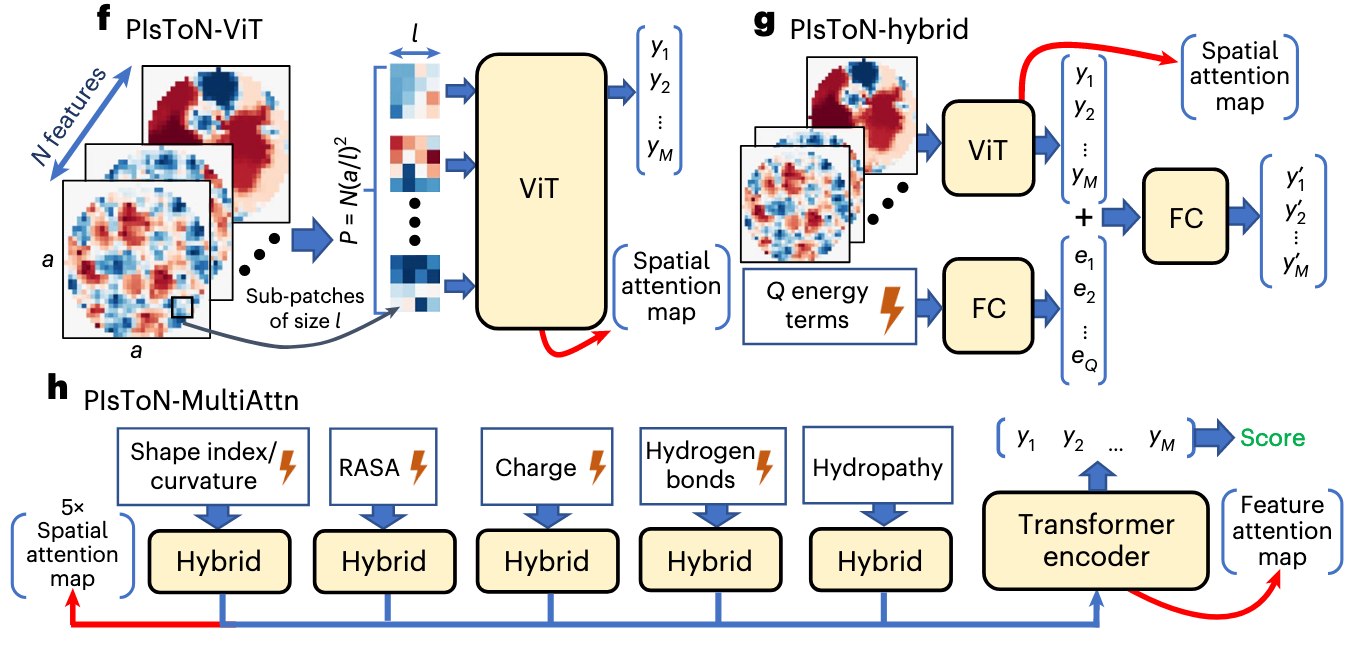
PIsToN-ViT
这一步采用了为图像分类开发的 ViT 架构。模型的输入是从蛋白质界面投影得的多个 特征图( ) ,共同组成的 通道特征图,维度为 。
- Sub-patches:用 的小正方形将特征图进行分割,产生 个小正方形,此时维度为 。
- Linear:将小正方形投影到一个 维的嵌入。
- Class Token:与传统的 ViT 做法一致,将数据的类标签也通过一个 投射到 Embedding 上。
- Position Embedding:作者在这里使用了可学习的位置编码。
- Output:最终经过将上述输入进行合并,经过多层自注意力,输出的是特征图的 维嵌入向量 。
上述流程用公式化表示为:
此时的注意力图称为空间注意力图,标识了对分类决策至关重要的 小正方形 Patch 。
PIsToN-hybrid
该架构在 PIsToN-ViT 的基础上,加入了基于物理先验的能量项,简单的使用 Linear 的全连接将 项能量项投射到嵌入向量 上,接着和从 ViT 获得的向量 进行拼接,再被一个 Linear 投射回嵌入维度,从而融合空间和物理信息。
PIsToN-MultiAttn
这部分实现了 多通道的注意力机制 ,其创新点在于将不同的特征图和能量项进行混合通过 PIsToN-hybrid 进行拼接,分五个分支独立地从不同空间和物理先验上学习起嵌入表征,这五组通道在 特征融合 一节就已经讲过。
这五个通道分别包含一个独立的 ViT-Hybrid 网络,获得了五组表征和空间注意力图,此时,为了防止类信息的遗忘,在输入最终融合五组表征的 Transformer 之前再次加入了 Class Token 。最终,五组表征和类信息输入到最终的 Transformer 网络中,得到了最终的嵌入表征 和特征注意力。
为什么最终输出的不是类别?—— 原型学习
事实上,对于结合界面是否是 native 的这个问题,本质上就是一个二分类的问题。我们完全可以对结合界面分类为是 native 的和不是 native 的,并使用 PIsToN-MultiAttn 模型进行预测,但作者通过实践告诉我们直接的二元分类并不能够很好的解决这个问题。其原因可以考虑这几个方面:
- 评分函数的核心任务不仅是判断“对”或“错”,更重要的是从成千上万个候选构象中将 最接近天然结构(Native-like)的构象排在最前面 。
- 传统的二元分类器(如仅使用交叉熵损失)往往只关注区分边界,无法为大量的候选构象提供足够细微的排序区分度。
这也就是这篇论文的创新之处,作者引入对比学习、原型学习的策略,PIsToN-MultiAttn 最终输出的是其类别的嵌入表征,通过距离分类中心的方式度量了构象质量的优劣和排名,从而对不同构象进行打分评估。
PIsToN 训练流程
对比学习
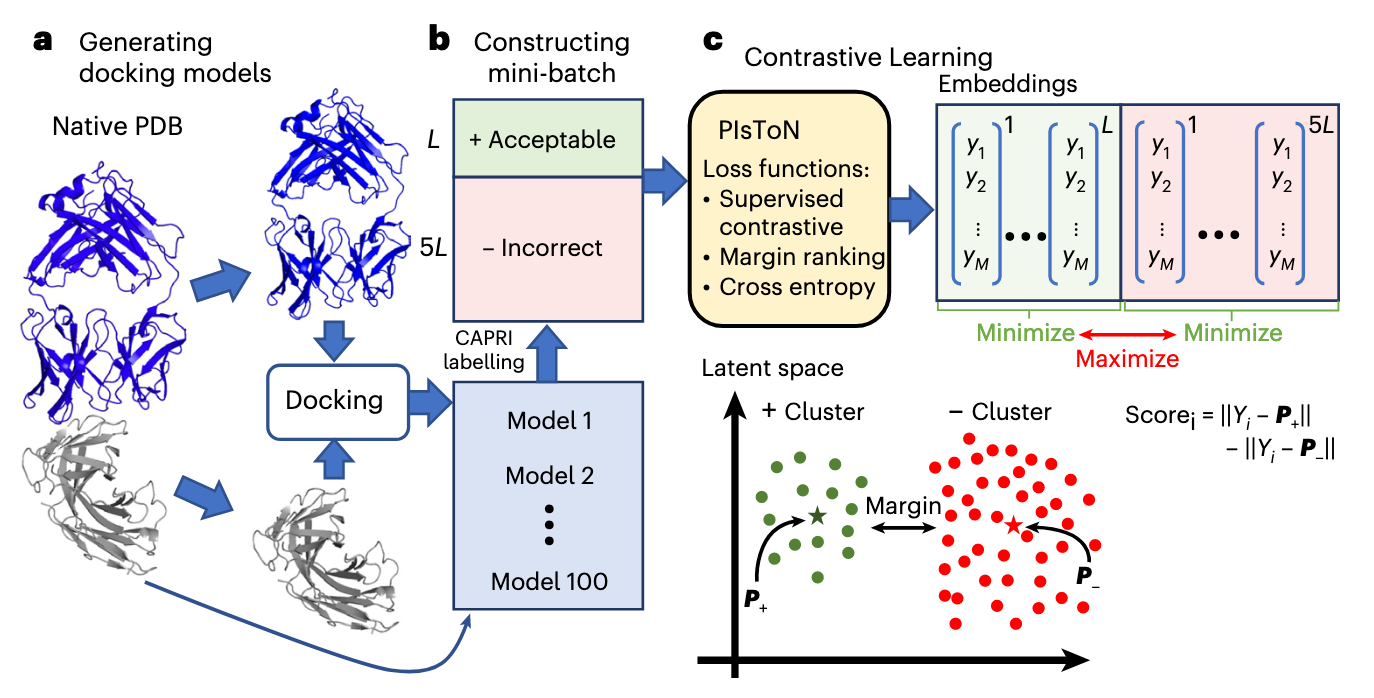
CAPRI 标准
该标准是传统方法用于评估对接模型质量的一个标准。
包含三个独立的指标:Fnat(保留的天然界面接触比例)、LRMSD(配体链的均方根偏差,反映整体结构对齐)、iRMSD(界面残基的均方根偏差,反映局部细节)。
这些指标需通过复杂阈值组合将模型分为四类:错误(Incorrect)、可接受(Acceptable)、中等(Medium)和高质量(High)。
为了实现对比学习的优化,数据集的构造需要构造不同的正负样本。从训练集中的天然蛋白质复合物(native)出发,使用 HDOCK 重新对接了 100 个对接 model。然后将每一个对接的 model 和 native 进行比较以通过 CAPRI 标准评估其质量。满足: 的认为是可接受的对接 model,否则是不正确的 model。
同时,作者设计了在同一个蛋白质下构造正负样本对,而不是随机构造的方法。经实验验证,这样的选取策略效果更好。每一个小批次包括同一个蛋白质的可接受和不正确 model,限制正负样本的比例为 1:5 以限制 model 中大量都是不正确的数据不平衡问题。除了进行输入之前的标准缩放的归一化操作,在训练过程中作者还对二维图进行 0~360° 随机旋转的数据增强,迫使训练的模型具有旋转不变性。
Loss
训练过程由对比学习指导,其损失函数包括三项:监督对比损失、边际排序损失、二元交叉熵损失。
Supervised Contrastive Loss
该损失有如下公式所计算,第一部分是对正样本做对比,让正样本之间相互靠近;第二部分是对负样本做对比,让负样本之间相互靠近。从而让 同类样本 在嵌入空间中聚在一起(拉近正样本距离),不同类样本 互相推开(拉远负样本距离)。
Margin Ranking Loss
该损失按照如下公式计算,用来学习两个聚类原型(正类原型 P+、负类原型 P−),让样本在嵌入空间里“归到自己类的原型,远离另一类原型”。
Binary Cross-Entropy Loss
二元交叉熵损失就是分类问题中最常用的交叉熵损失,直接优化分类的准确:
实验
Datasets
MaSIF
作者使用 MaSIF 工作中的数据来进行训练和测试,预处理了 4,942 个复合物训练、181 个复合物验证(验证集是从测试集中抽取 20% 获得的)和 678 个天然蛋白质复合物测试。总共生成了 17291 个正样本和 452031 个负样本用于训练集、703 个正样本和 16864 个负样本用于验证集、随机选择了一个正样本和一个负样本共产生了 678 个正样本和负样本用于测试。
CAPRI-score
来自 CAPRI 挑战集的13对蛋白质用于测试排名性能。作者使用来自 DeepRank 研究的蛋白质对接模型。数据集包括由 HADDOCK 对接工具生成的 16581 个复合物以及相应的 CAPRI 质量标签用于测试。
PDB-2023
作者选择 2023 年加入到 RCSB 组织维护的 PDB 中的蛋白质复合物。其目的是最大限度地减少来自所有工具的训练集的偏差。作者处理得到 53 个非冗余的异源二聚体,接着每个天然复合物的蛋白质链被分离并使用 HDOCK 重新对接,最终得到了 126 个可接受 model 和 5174 个不正确 model 作为测试集。
Lab1:PIsToN 模型的确定
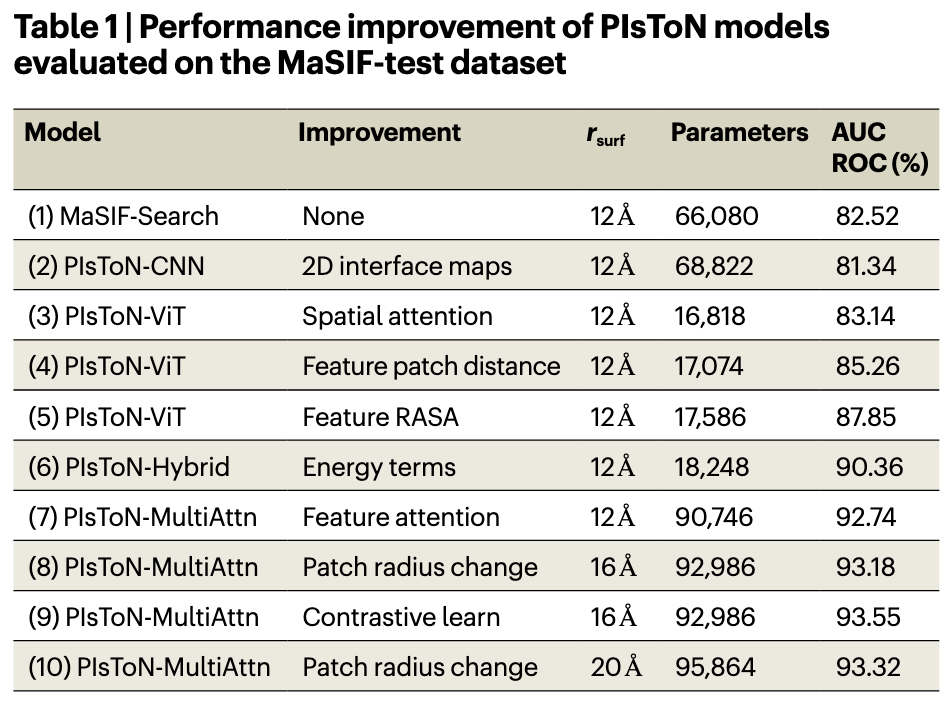
- MaSIF、CNN 作为 Baseline,融合 Transformer 的注意力模型预测效果更好。
- Multi-Attention 、Hybrid 机制的融入增强了预测效果。
- 作者有尝试不同的 ,效果改变不大,为减少计算,选用 9 号模型。
Lab2:Datasets 的测试结果
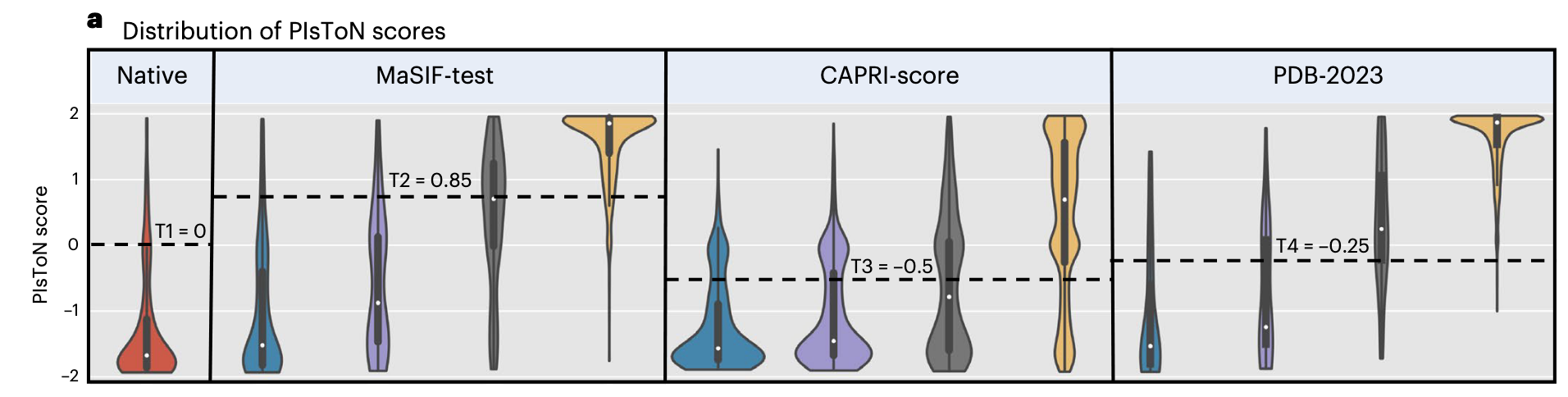
- 结合预测的打分就是距离两个原型之间的距离: 。
- 作者计算了不同的 阈值和马修斯相关系数,选取最大马修斯相关系数对应的阈值作为分类阈值。
Lab3:PIsToN 的可解释性机制
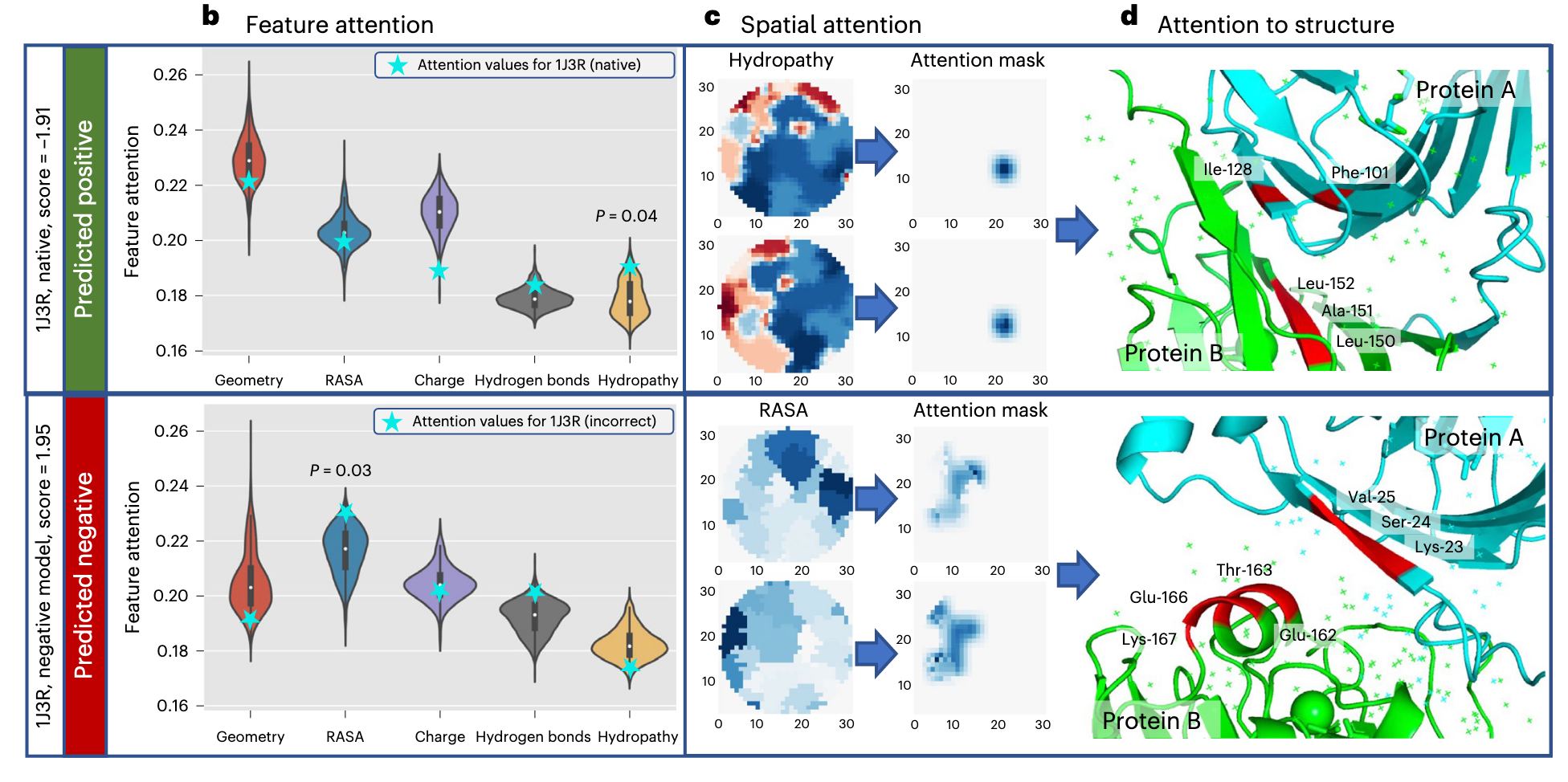
- PIsToN 输出特征注意力图和空间注意力图,正样本更关注集合特征,而负样本更关注相对可接受表面积。
- 作者探究了一个实际的例子:来自考古热球菌litoralis50的蛋白质磷酸粒糖异构酶。发现在 native 的结构上其对疏水性的空间注意力和蛋白质界面上的残基相对应。同时对其 HDOCK 出的不正确 model 界面上预测,其 RASA 的空间注意力分数表明:突出显示的残基在两种蛋白质上都是表面可访问的,但太远了,可能会破坏该位置可能存在的任何复合物的稳定性。
Lab4:不同打分工具性能的比较
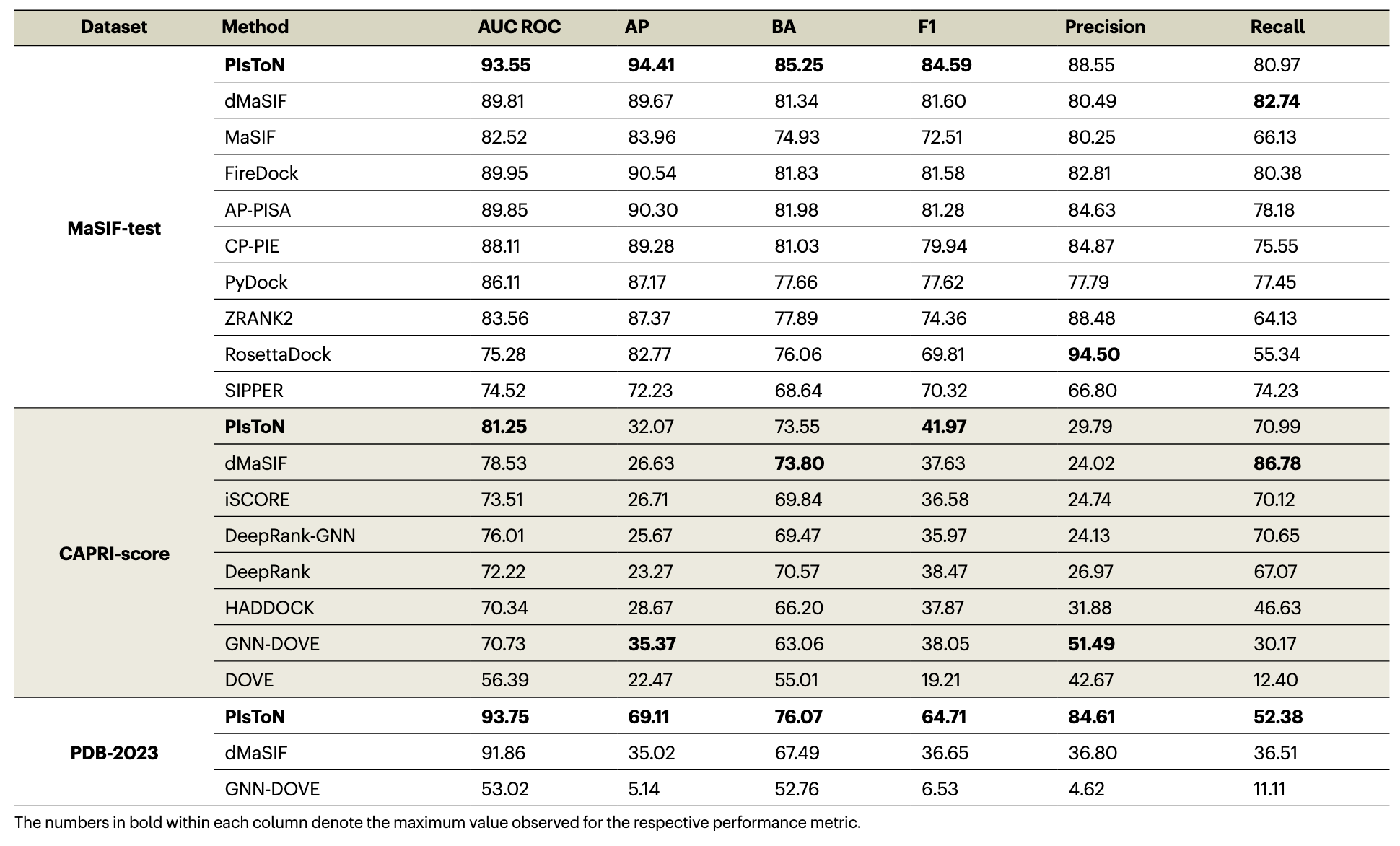
- PIsToN 在三个数据集上的大部分分类指标都较好。
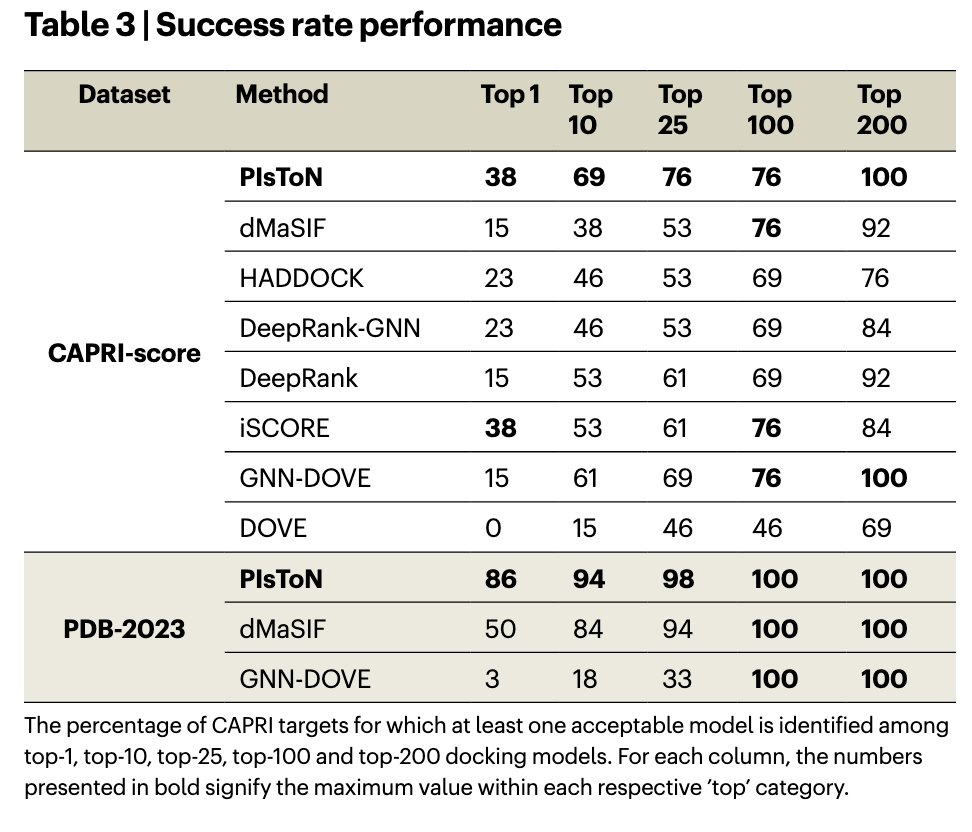
- 成功率分析表明,PIsToN 在对 CAPRI 分数和 PDB-2023 数据集的许多生成模型中的对接姿势进行排名的任务中表现更好。
Lab5:运行时间比较
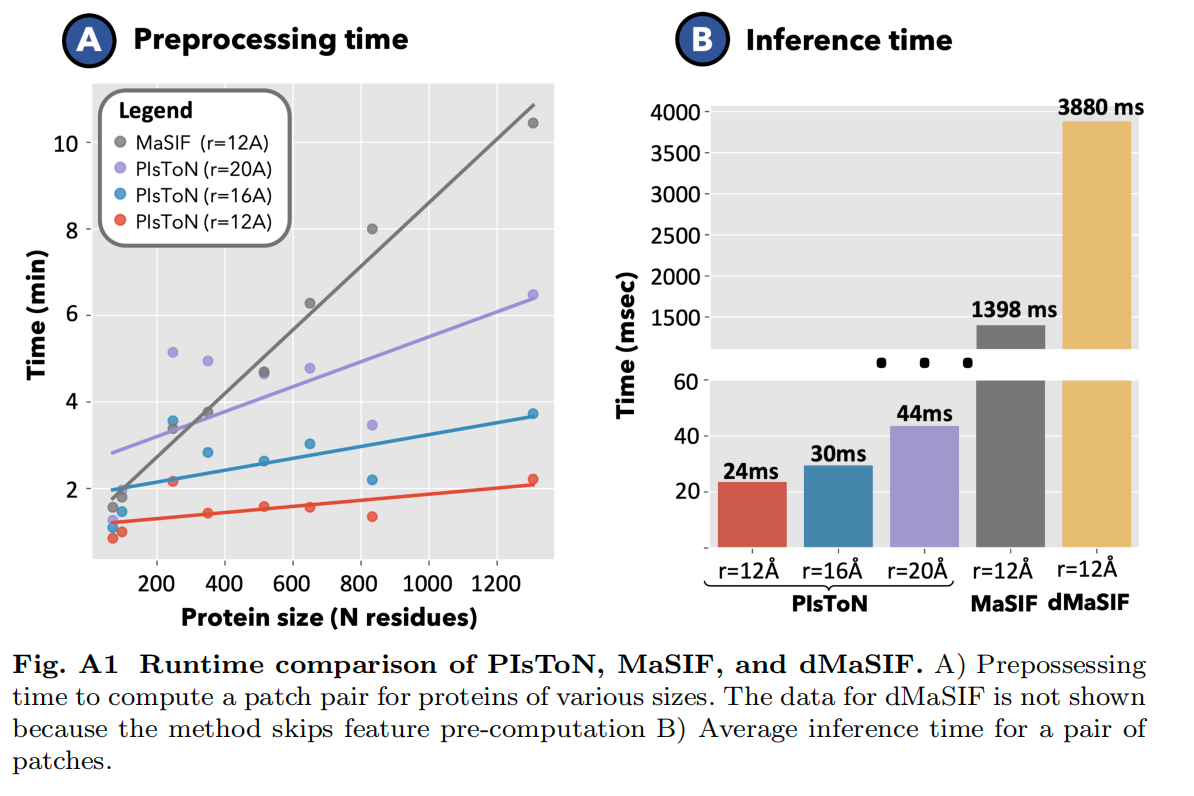
- PIsToN 预处理计算效率高,且推理时间快。
相关工作
 PUMBA
PUMBA结语
这篇文章详细讲解了《Evaluating protein binding interfaces with transformer networks》这篇文章,主要用于个人研究和记录。